DNAML und ProML
DNAML und ProML sind zwei weitere Programme aus der Phylib Programmsammlung von Joseph Felsenstein.
Sie benutzen beide Maximum Likelihood-Methoden, um Verwandschaftsbeziehungen zwischen verschiedenen genetischen oder Proteinsequenzen zu bestimmen.
Mit dieser Maximum Likelihood-Methode versucht das Programm nun mit vorgegebenen Parametern die Wahrscheinlichkeit zu errechnen, dass die eine Sequenz in die andere mutiert sein könnte und erstellt anhand dieser Wahscheinlichkeiten einen Baum.
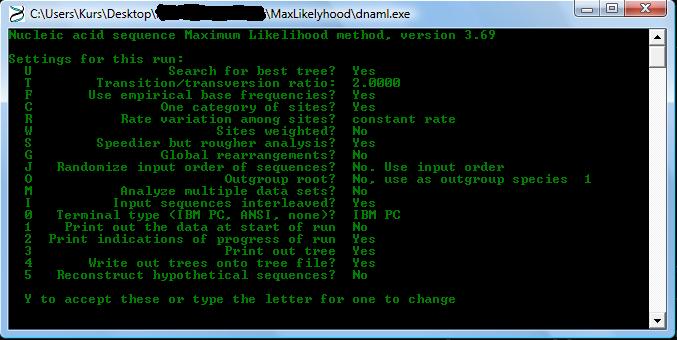 |
| Abb.1.: Hier sieht man DNAML ausgeführt |
Wie zu sehen ist gibt es hier sehr viele Möglichkeiten die Suche zu individualisieren. Die Wichtigste hiervon ist jedoch die Variation der Transition:Transversions Rate. Bei diesem Wert kann man einstellen, wie hoch die Wahrscheinlichkeit einer Transversion im Vergleich zu einer Transition ist. Obwohl es mehr Möglichkeiten für eine Transversion (Austausch einer Purin durch eine Pyrimidinbase) gibt finden diese deutlich seltener statt, als Transversionen (z.B. Austausch einer Purin durch eine andere Purinbase). Dieser Wert lässt sich nicht gut bestimmen, so daß man entweder in empirischen Reihen bestimmte Werte nimmt, oder einfach eine beliebige Zahl zwischen 2-10 nimmt, was dar stellt, dass Transversionen deutlich unwahrscheinlicher sind.
Das dies natürlich zu Ungenauigkeiten führt ist verständlich.
Wichtige Vorbedingungen von DNAML sind, dass sämtliche Basen gleich behandelt werden, was in der Natur ja nicht unbedingt der Fall ist und dass bei jeder Mutation auch z.B. ein A durch ein A ersetzt werden kann, so dass sich eigentlich nichts ändert.
Die Wahrscheinlichkeit, der verschiedenen Mutationen ist in den Standardeinstellungen durch das empirische Vorkommen der Basen in dem Strang definiert, kann aber jedoch auch manuell eingegeben werden.
Wenn nun all diese Wahrscheinlichkeiten für die Einzelergebnisse gegeben sind versucht das Programm nun damit den Stammbaum zu finden, der die höchste Wahrscheinlichkeit besitzt.
Genauere Erklärungen zu den verschiedenen Befehlen gibt es in der Dokumentation.
ProML
 |
| Abb.2.: Hier sieht man ProML ausgeführt |
ProML ist eine Abwandlung von DNAML, welches auch mit der Maximum Likelihood-Methode nach Stammbäumen für verschiedene Aminosäure-Sequenzen sucht. Hierbei orientiert es sich an verschiedenen Matrizen, für Aminosäureaustauschwahrscheinlichkeiten. Namentlich die Jones-Taylor-Thornton, PAM oder PMB-Matrizen. Diese beinhalten die Wahrscheinlichkeit für jede Aminosäure in eine andere umgetauscht zu werden, anhand ihrer chemischen Eigenschaften und empirisch belegter Werte.
Somit entfällt hier schon mal das selbstständige Festlegen der Wahrscheinlichkeiten, was dieses Programm fehlerunanfälliger macht.
Erwähnenswert ist noch, dass man mit dem Programm die Möglichkeit hat verschiedene Abschnitte des Stranges, mit der Weimit verschiedenen Mutationswahrscheinlichkeiten zu versehen, so dass zum Beispiel das aktive
Sonst ähneln die Optionen größtenteils denen anderer Phylib Programme und werden alle in der Dokumentation geklärt.
Beide diese Programme sind in der Phylib Sammlung zu finden, welche von Joseph Felsenstein veröffentlicht wurde.